Neurobiologie moléculaire et cellulaire de C. elegans
Chef d'équipe: Thomas Boulin — Site de l'équipe
C. elegans | excitabilité cellulaire | canaux potassiques | génetique | neurobiologie moléculaire et cellulaire | édition dirigée du génome par CRISPR/Cas9
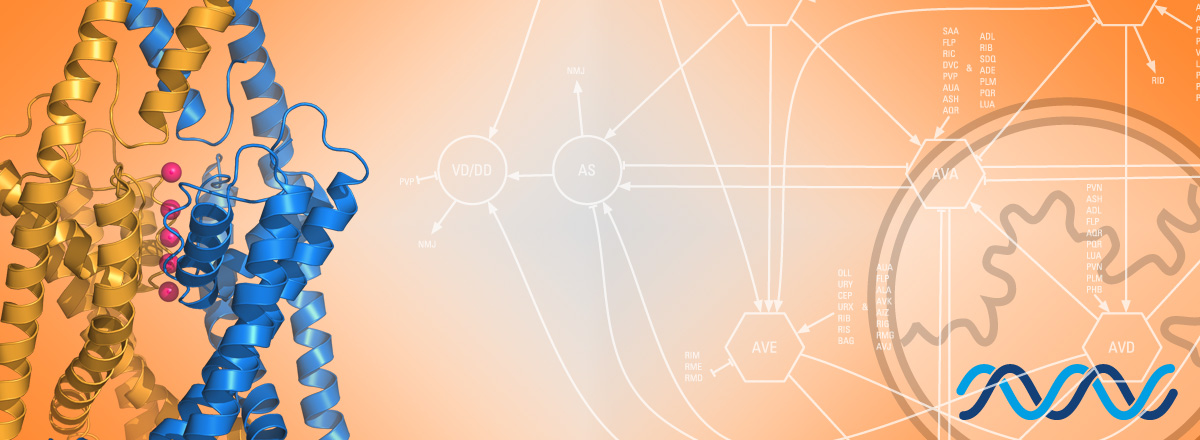
Les canaux potassiques jouent un rôle central dans l’établissement du potentiel de membrane des cellules animales. Nous utilisons des approches génétiques chez le nématode modèle C. elegans afin d’élucider les mécanismes moléculaires et cellulaires qui contrôlent le nombre, l’activité et la localisation subcellulaire des canaux potassiques à deux domaines pore (K2P).
New publication in The Journal of Physiology - Caenorhabditis elegans as an in vivo model system for human inherited primary arrhythmia syndromes
2025-12-05
Congratulations to Antoine for this expansive
review!.
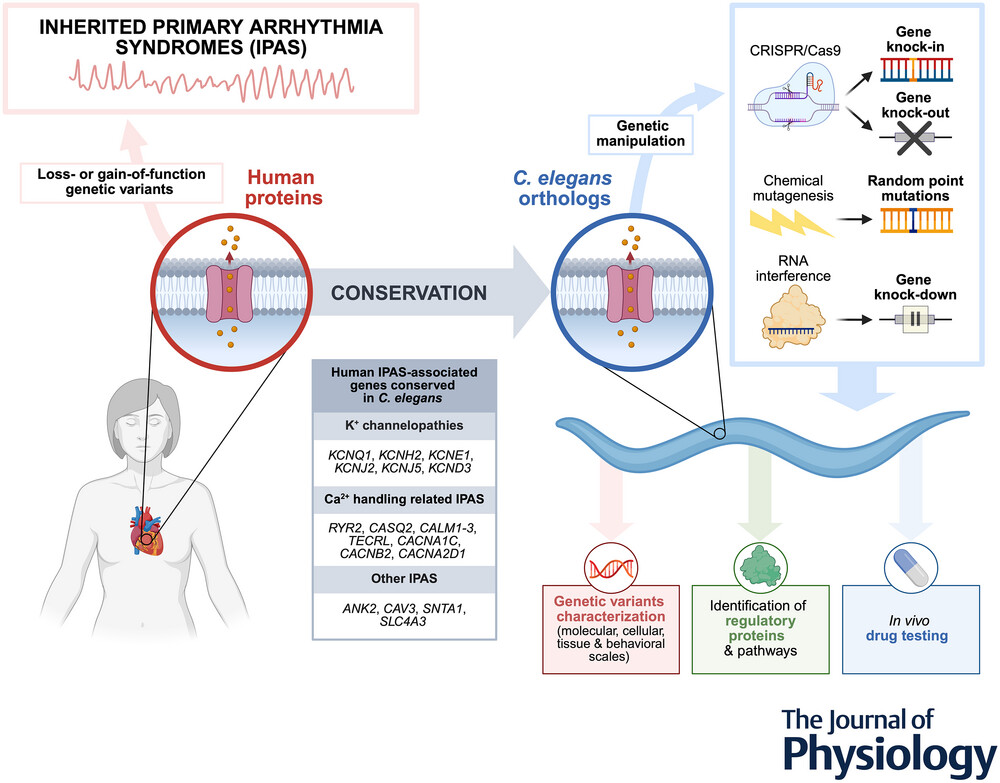
Inherited primary arrhythmia syndromes (IPAS) are genetic heart diseases associated with an elevated risk of sudden cardiac death, particularly in young individuals. Modelling these rare and serious conditions is essential to elucidate their mechanisms and to identify new treatments.
Most genes involved in IPAS (e.g., congenital long-QT syndrome, catecholaminergic polymorphic ventricular tachycardia, calcium-release deficiency syndrome, Andersen-Tawil syndrome, Timothy syndrome, calmodulinopathies, and short-QT syndrome) are conserved in Caenorhabditis elegans, a model organism that offers powerful genetic tools for precise gene manipulation, including knock-in, knock-out, and knock-down approaches. In vivo studies in C. elegans can be used to characterize the consequences of genetic variants (at molecular, cellular, tissue, and behavioural scales), to identify new regulatory proteins, and to perform drug testing.
Here we summarize the characteristics of human IPAS and highlight the accumulating evidence that supports the utility of C. elegans as a simple yet powerful in vivo model for these diseases, capable of filling the gap between in vitro studies and complex transgenic animal models.
New publication in PNAS - Constitutive sodium permeability in a C. elegans two-pore domain potassium channel.
2024-10-15

Congratulations to all involved in this multi-disciplinary exploration of
the molecular determinants of K2P channel selectivity.
Potassium channels play a central role in modulating cellular excitability, particularly of neuronal cells. Their unique structure determines their ability to let ions pass selectively through cell membranes. The impact of pathological or evolutionary variations in this selectivity filter remains difficult to predict. Here, we reveal that UNC-58, a member of the two-pore domain potassium (K2P) channel family of C. elegans, exhibits an unusual sodium permeability due to a unique cysteine residue in its selectivity filter. Our findings underscore the importance of functional studies to determine how sequence variation in potassium channel selectivity filters can shape the electrical profiles of excitable cells.
New publication - Wnt-Ror-Dvl signalling and the dystrophin complex organize planar-polarized membrane compartments in C. elegans muscles
2024-06-10

Our study revealing the remarkably complex organisation of the worm's sarcolemma is now out at
Nature Communications! Almost six years in the making, congratulations to Alice, Nora, Marie, Amandine, Noémie, Elise and Olga for this
magnum opus revealing this entirely unsuspected new case of
planar cell polarity in
C. elegans muscles.
SUMMARY
Cell polarity mechanisms allow the formation of specialized membrane domains with unique protein compositions, signalling properties, and functional characteristics. By analyzing the localization of potassium channels and proteins belonging to the dystrophin-associated protein complex, we reveal the existence of distinct planar-polarized membrane compartments at the surface of C. elegans muscle cells. We find that muscle polarity is controlled by a non-canonical Wnt signalling cascade involving the ligand EGL-20/Wnt, the receptor CAM-1/Ror, and the intracellular effector DSH-1/Dishevelled. Interestingly, classical planar cell polarity proteins are not required for this process. Using time-resolved protein degradation, we demonstrate that –while it is essentially in place by the end of embryogenesis– muscle polarity is a dynamic state, requiring continued presence of DSH-1 throughout post-embryonic life. Our results reveal the unsuspected complexity of the C. elegans muscle membrane and establish a genetically tractable model system to study cellular polarity and membrane compartmentalization in vivo.
New publication - Functional and clinical characterization of a novel homozygous KCNH2 missense variant in the pore region of Kv11.1 leading to a viable but severe long-QT syndrome
2023-12-21

Congratulations to Antoine, Olga and the team of
Philippe Chevalier (Rhythmology department, HCL Lyon) for our first collaborative study about
Kv11.1/hERG potassium channels.
Highlights
- Homozygous missense variants in the pore of Kv11.1 often lead to intrauterine death.
- We describe a novel hERG p.Gly603Ser homozygous loss-of-function variant.
- It causes a severe but viable long-QT syndrome with a delayed clinical expression.
- Family segregation and functional analysis classify hERG p.Gly603Ser as probably pathogenic.
DYSCO - Dystrophin-associated protein complex and subcellular compartmentalization
2023-07-13

Our project with the team of
Vincent Mirouse (iGred, Clermont-Ferrand) and
Helge Amthor (UVSQ) has been selected by ANR ! The Dystrophin Associated Protein Complex (DAPC) is a key actor of the cell – extracellular matrix (ECM) interface, as revealed by its implication in human genetic disorders. However, its molecular and cellular functions are still poorly understood because tractable model systems allowing state-of-the-art in vivo cell biology and genetic approaches are lacking. Our three teams have developed the first transgenic dystrophin reporters that have revealed remarkably compartmentalized membrane distributions of the DAPC in epithelia and muscle cells in
C. elegans, Drosophila, and mouse. The project aims to elucidate the organization and dynamics of the DAPC and to characterize its new functions in relation to specific cell cortical compartments.
Fleggsibility - Dissecting the microevolutionary flexibility of a neural circuit
2022-08-13
Our project with the team of
Christian Braendle (iBV Nice) and
ViewPoint: Behavior Analysis Technologies has been selected by ANR ! On the heals of our joint publication (Vigne et al.,
Science Advances 2021) we will be trying to understand how specific neural circuits evolve to generate natural behavioural variation within species. In particular, the precise genetic changes that modulate cellular and developmental architectures of reproductive systems remain unclear. Here we will focus on the simple egg-laying circuit of the nematode
Caenorhabditis elegans as a powerful model system to study natural microevolutionary (intraspecific) variability.
Join the team
We are always looking for highly motivated Post-docs, Masters and PhD students to participate in our projects. Please consult
this page to apply L’établissement et le maintien d’un potentiel électrique de membrane négatif sont essentiels au bon fonctionnement de la plupart des cellules et en particulier des cellules excitables. Les canaux potassiques de la famille K2P (two-pore domain potassium channels) jouent un rôle prépondérant dans ce processus. Ces canaux décrits pour la première fois en 1995 sont largement conservés au cours de l’évolution. Ils sont exprimés dans de nombreux types cellulaires et ont été impliqués dans des fonctions physiologiques diverses, tel que la régulation de l’activité neuronale, respiratoire et cardiaque, ou le contrôle de la prolifération cellulaire et de la sécrétion hormonale. Les canaux K2P sont des cibles principales des anesthésiques généraux. L’activation des K2Ps explique en grande partie l’effet immobilisant et sédatif de certains anesthésiques. Des mutations de canaux K2P chez l’homme conduisent à des pathologies cardiaques (Friedrich, 2014), et au syndrome de Birk Barel, une maladie génétique rare caractérisée par un retard mental, une hypotonie et des déformations caractéristiques de la face (Barel, 2008).
Malgré la diversité des processus physiologiques, pathologiques et développementaux impliquant les K2Ps, les gènes et les mécanismes cellulaires qui contrôlent la biologie cellulaire et l’activité de ces canaux sont encore mal connus. Notre équipe utilise une approche génétique chez le nématode modèle Caenorhabditis elegans afin d’identifier de nouveaux facteurs et les mécanismes cellulaires qui contrôlent le nombre, l’activité et la distribution subcellulaire des canaux potassiques à deux domaines P. Nous employons toute la gamme des techniques disponibles chez C. elegans, tel que la génétique, l’imagerie in vivo, l’électrophysiologie, ou les technologies les plus récentes permettant la modification ciblée et le séquençage complet des génomes. Ces études permettront d’identifier de nouvelles voies de régulation des K2Ps et de mieux appréhender les mécanismes qui régulent l’activité de ces canaux dans d’autres organismes.
Two-pore domain potassium channels (K2P) play a central role in the control of cellular excitability and the regulation of the cell's electrical membrane potential. K2Ps have been widely conserved throughout evolution. They are polymodal ion channels that are subjected to extensive regulation by a diverse set of physical (pH, temperature, mechanical force) and biological signals (lipids, G-protein coupled receptor pathways). They are broadly expressed in excitable and non-excitable cells, and have in turn been implicated in a large spectrum of physiopathological processes, ranging from the regulation of neuronal excitability, respiratory and cardiac function to the control of cell volume, hormone secretion and cell proliferation. Recently, loss- and gain-of-function mutations in K2P channels have been directly linked to human pathologies (Birk Barel syndrome, familial migraine with aura, cardiac conduction disorder, FHEIG neuro-developmental disorder).
In contrast to many other ion channel families, comparatively little is known about the molecular and cellular processes that regulate different aspects of the cell biology of K2P channels. For instance we know only of very few factors that specifically regulate the expression, the activity and the localisation of K2P channels at the cell surface. Therefore the central question addressed by our team is: How is the number of active two-pore domain potassium channels present at the cell surface controlled in vivo?
To identify novel genes and conserved cellular processes that regulate the biology of K2P channels in vivo we take advantage of the powerful genetic tools available in the model nematode Caenorhabditis elegans. We use the full array of techniques available in C. elegans including genetics, live imaging, electrophysiology and state-of-the-art CRISPR/Cas9 genome engineering and next-generation DNA sequencing. These studies will provide new leads to understand the cellular pathways that control K2P function in other organisms.
The starting point for our genetic screens are gain-of-function mutants of K2P channels that cause strong and easily identifiable behavioral phenotypes. This approach has become possible because of a recent discovery of our group. By combining electrophysiology in Xenopus oocyte and CRISPR/Cas9-based gene editing in C. elegans, we have shown that a single conserved residue controls the activity of all K2P channels from vertebrates and invertebrates (Ben Soussia et al., Nature Communications 2019). This has allowed us to rationally design novel gain-of-function mutants for C. elegans K2P channels and to identify genes that are specifically required to control their expression, biogenesis, trafficking, and subcellular distribution.
Rationale
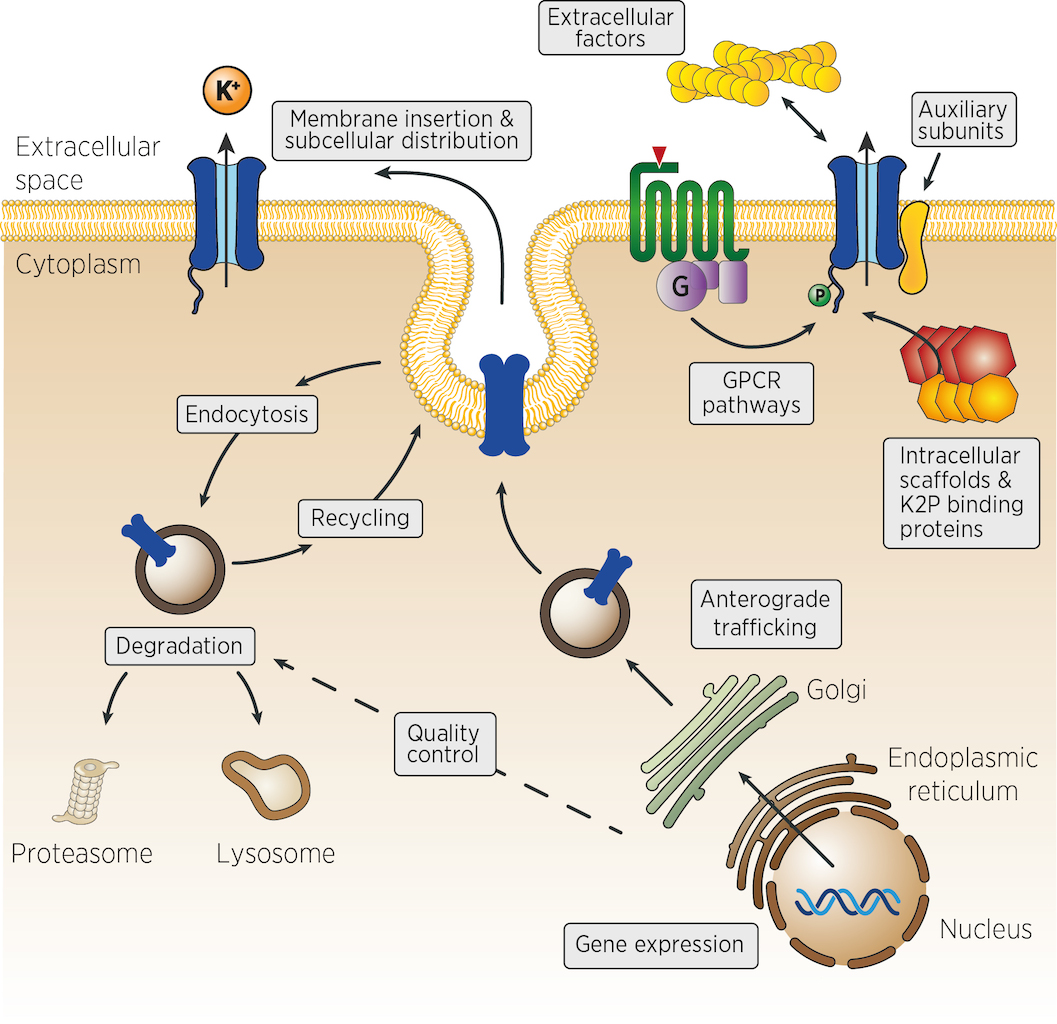
Many different mechanisms could control the number, the activity and the localisation of K2P channels in vivo. From the control of channel expression, biosynthesis and trafficking, to the regulation of membrane insertion, recycling and channel activity at the cell surface (figure). Very few experimental strategies can identify, at once, genes acting in such diverse aspects of cellular physiology. We propose that genetic strategies could address many of these levels of regulation.
Genetic approaches have been vastly underused by past research focusing on K2P channels. Yet, genetic screens in model organisms such as flies and worms are ideally suited to dissect complex cellular processes and gene regulatory networks in vivo. C. elegans has proven a powerful, integrated model system for the study of synaptic function, development, aging, cell death or gene regulation by microRNAs and RNA interference. Due to its short life cycle (< 3 days), C. elegans is well suited for large-scale genetic screens. In addition, most mutants of neuronal genes are viable, and even paralyzed worms are able to develop and reproduce.
Recent estimates show that 40% to 75% of human disease genes have homologues in C. elegans. Conversely, about 38 % of the ~21,000 predicted genes of C. elegans have clear homologues in humans (Culetto and Sattelle, 2000; Silverman et al., 2009; Shaye and Greenwald, 2011). Because of this evolutionary conservation, the genes or pathways that regulate K2P function in C. elegans should enable us to identify genes or mechanisms that are also relevant to the regulation of vertebrate K2P channels in excitable and non excitable cells.
Questions that we would like to solve
- What are the cellular mechanisms that control the distribution of K2P channels in vivo in C. elegans?
- Are these pathways conserved in other organisms, including vertebrates?
- How are the number and the activity of K2P channels at the cell surface controlled?
- Are there specific factors that differentially regulate K2P channels in distinct tissues or cell types?
- Do regulators of K2P channels also control the biology of other ion channel families?
Genetic regulation of K2P channels expression in C. elegans
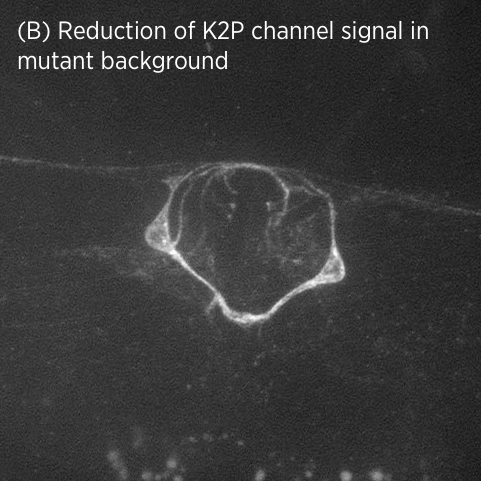
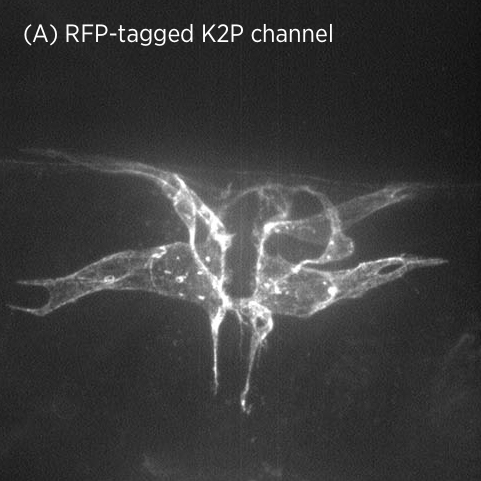 Using CRISPR/Cas9 genome engineering, we have generated the first knock-in line of a two-pore domain potassium channel. This fluorescently-labeled ion channel is fully functional and visible in vulval muscles and VC motoneurons associated with the vulval muscles and egg-laying system (A).
Using CRISPR/Cas9 genome engineering, we have generated the first knock-in line of a two-pore domain potassium channel. This fluorescently-labeled ion channel is fully functional and visible in vulval muscles and VC motoneurons associated with the vulval muscles and egg-laying system (A).
In mutant strains isolated in our genetic screen, K2P channel expression is lost specifically in vulval muscles but not in neurons (B). This suggests that K2P channel expression may be differentially regulated in neurons and muscle.
The regulatory genes we identified belong to a well-known signalling cascade and include a ligand, receptor, processing enzyme and transcription factor. Identifying an entire genetic pathway in a single forward genetic screen illustrates the power of these genetic approaches.
Subcellular localisation of K2P channels
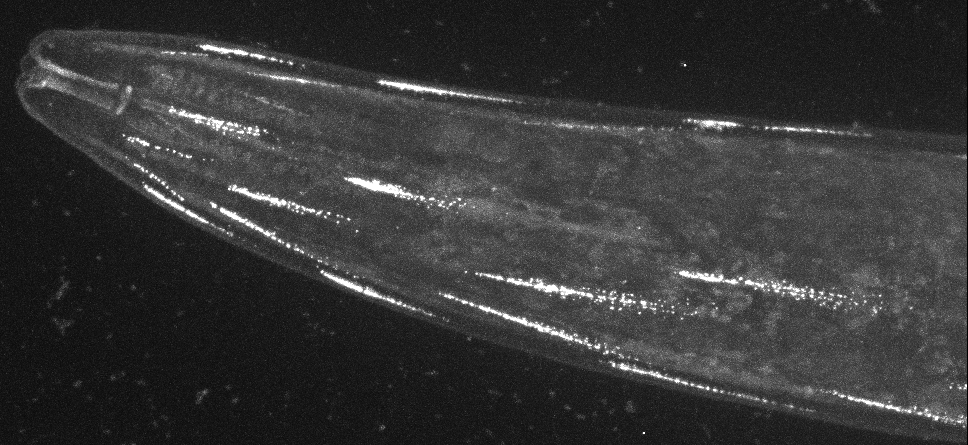 Direct visualisation of a K2P channel in vivo and at physiological expression levels opens the way to further analyse the cellular and molecular pathways that regulate the biology of these important ion channels. For example, CRISPR-based labeling of the TWK-28 channel revealed and unexpected and fascinating subcellular localisation pattern. TWK-28 channels are found at the anterior tip of individual muscle cells, revealing an entirely unforeseen asymmetry in the organisation of the C. elegans muscle membrane. Using a forward genetic screen targeting regulators of TWK-28, we discovered a novel link to the Dystrophin-Associated Protein Complex (DAPC).
Direct visualisation of a K2P channel in vivo and at physiological expression levels opens the way to further analyse the cellular and molecular pathways that regulate the biology of these important ion channels. For example, CRISPR-based labeling of the TWK-28 channel revealed and unexpected and fascinating subcellular localisation pattern. TWK-28 channels are found at the anterior tip of individual muscle cells, revealing an entirely unforeseen asymmetry in the organisation of the C. elegans muscle membrane. Using a forward genetic screen targeting regulators of TWK-28, we discovered a novel link to the Dystrophin-Associated Protein Complex (DAPC).
The Dystrophin-Associated Protein Complex (DAPC) is a major actor of the relationship between cells and their extracellular matrix. Because this complex can physically connect the actin cytoskeleton and ECM proteins, it is generally assumed that its main function is to ensure Cell cytoskeleton-ECM cohesiveness along muscle sarcolemma. This view is still prevalent to explain the pathogenesis of Duchenne Muscular Dystrophy and other muscle diseases resulting from DAPC disruption in patients. Despite the significant progress made regarding the clinical and genetic dimensions of Dystrophin-dependent physiopathology, the precise molecular, cellular, and developmental functions of the DAPC are not yet fully understood. As part of a project funded by AFM Téléthon (Alliance MyoNeurALP), our group is using state-of-the art genetic and imaging tools in the model nematode C. elegans to challenge the simplistic view of a predominantly mechanical role of the Dystrophin-associated protein complex. In fact, our results suggest that the DAPC may be at the heart of cellular mechanisms that precisely define subcellular cortical domains allowing membrane proteins to be compartmentalized within a single cell.
Indeed, by systematically analyzing the subcellular distribution of membrane proteins and ion channels using CRISPR/Cas9 gene editing, we have discovered that specific subcellular compartments are constructed at the surface of muscle cells by the Dystrophin-associated complex. In turn, ion channels and neurotransmitter transporters are recruited to these distinct subcompartments, likely providing them with specific electrophysiological properties.
This striking example of membrane compartmentalization was entirely unsuspected and raises a number of exciting questions.
Molecular, cellular, and clinical investigation of the autism, epilepsy, and neurodevelopmental disorder gene Neurobeachin/NBEA
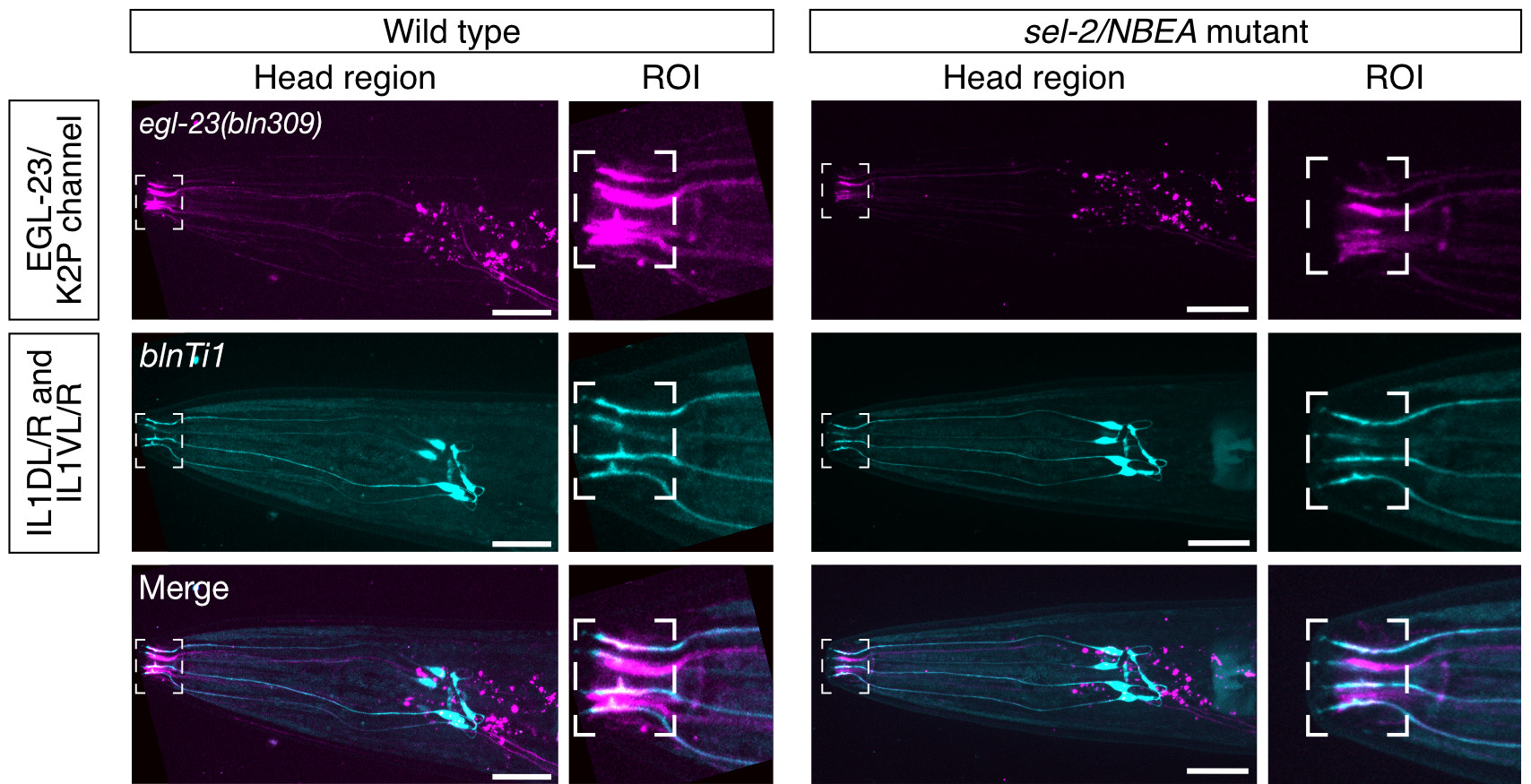 Neurobeachin is a brain-specific protein required for vesicular trafficking, synaptic structure, and synaptic targeting of neurotransmitter receptors. Neurobeachin mutations have very recently been identified as a genetic cause of autism, neurodevelopmental delay and early generalized epilepsy, now known as NEDEGE (Mulhern et al., 2018).
Neurobeachin is a brain-specific protein required for vesicular trafficking, synaptic structure, and synaptic targeting of neurotransmitter receptors. Neurobeachin mutations have very recently been identified as a genetic cause of autism, neurodevelopmental delay and early generalized epilepsy, now known as NEDEGE (Mulhern et al., 2018).
Many of the 24 cases described in this cohort were still young children at the time they were reported. Thus, we currently only have a very limited understanding of the range of human disease phenotypes and the natural history of NBEA encephalopathy. It is therefore essential to pursue in-depth clinical analysis based on neuropsychological evaluations and electroencephalography to delineate in richer breadth and depth the clinical expression of this recently discovered neurological disease.
While most reported probands carry strong loss-of-function alleles, single point mutations in conserved Neurobeachin residues are also found. Determining pathogenicity of these missense variants poses a major challenge, because there are presently no model systems, either cell-based or using genetic model organisms, in which single amino acid changes in Neurobeachin can be quickly generated and characterised in their native cellular context, limiting our understanding of genotype-phenotype relationships.
In a collaboration with the NIH’s Undiagnosed Diseases Network, we have for the first time used state-of-the-art gene editing and novel functional assays in the nematode Caenorhabditis elegans, to confirm the molecular diagnosis of a patient carrying a de novo variant (Boulin et al., 2021). Indeed, we demonstrate that mutating this single conserved residue has the same effect as a complete knock-out of Neurobeachin in the worm. Our findings thus open the way to study the functional impact of human genetic variants of Neurobeachin using the powerful genetic tools available in C. elegans.
In this project, we propose to combine clinical and fundamental research approaches to better understand the pathophysiology of this new neurological disease, identify cellular processes regulated by Neurobeachin, and find new molecular partners that could serve as potential therapeutic targets in the future.
This project is a collaborative venture between our team, Tristan T. Sands and Natalie Lippa (Columbia University, New York), Sarah Weckhuysen (VIB & University of Antwerp), Qiang Liu (Bargmann Lab, Rockefeller University, New York), and Maëlle Jospin (Bessereau Lab, Institut NeuroMyoGène, Lyon).



